

Draft Annex 11 Revision: From Interpretive to Prescriptive Regulation
The updates of EU / PIC/S Annex 11 and applicable sections of Chapter 4 represent a major change.

Introduction
EU good manufacturing practice (GMP) Annex 11 regulation on computerized systems has been effective since 1992.1 Following the data falsification uncovered at Able Laboratories in 2005,2 many data integrity (DI) issues involving computerized systems were uncovered during inspections.
In 2008, a proposed update to Annex 11 to reinforce DI controls was issued for industry comment.3 The revised version was issued in 20114 along with an updated EU GMP Chapter 4 on documentation.5 EU Annex 11 and Chapter 4 are also part of Pharmaceutical Inspection Convention Scheme (PIC/S) GMP, that are applicable in countries such as Japan, South Korea, Australia, New Zealand, etc.
In 2022, a concept paper on the revision of Annex 11 was issued by the European Medicines Agency (EMA) and PIC/S for industry comment,6 and in July 2025, the revised versions of Annex 11 and Chapter 4 were issued for stakeholder comments.7 At the same time, a new Annex 22 on artificial intelligence (AI) was also issued for stakeholder comment.7 We will not be discussing Annex 22 here.
This article provides detailed analyses of Annex 11 revision and the applicable clauses of Chapter 4 that impact computerized systems. The detailed discussion in this article includes:
- Why discuss Chapter 4 on documentation?
- Regulations: black, white or fifty shades of grey?
- Concept paper on the revision of annex 11
- Gone, but not forgotten?
- Regulatory plagiarism?
- Analysis and discussion of key sections of New Annex 11
- Chapter 4 clauses impacting computerized systems
- Hybrid systems
- Glossaries: problems, omissions and inconsistencies
- Summary
One of the major problems with the revision is that it is not easy to map to the current version of the regulation. Sections appear to have been selected by pulling names out of a hat.
Why discuss Chapter 4 on documentation?
As noted above, Annex 11 and Chapter 4 are revised in parallel. The rationale is:
To Fully Understand Annex 11, You Must Understand Chapter 4.
What is the rationale for needing to understand the implications of Chapter 4 on Annex 11? The clue is in the chapter title: Documentation. This includes risk management, electronic records, plus the underlying data/metadata, DI, data governance, ALCOA++ criteria, master documents, hybrid systems, signatures on GMP documents and record retention.
, Chapter 4 (documentation), Annex 15 (qualification and validation) and Chapter 7 (outsourced activities). Credit: Bob McDowall.")
Figure 1: Relationships between Annex 11 (computerized systems), Chapter 4 (documentation), Annex 15 (qualification and validation) and Chapter 7 (outsourced activities). Credit: Bob McDowall.
Considering the two regulations separately means that you miss the big picture shown in Figure 1. Therefore, this critical analysis of the revised Annex 11 will include appropriate sections of Chapter 4 as computerized systems create, interpret, report and store regulatory documents, records and data. Sections of both documents not discussed are shown in red in Figure 1. Readers must also be aware that there are requirements in Annex 15 on qualification and validation8 and Chapter 7 on outsourced activities9 that impact both Annex 11 and Chapter 4, also shown in Figure 1. We will only discuss relevant clauses where applicable. However, there is a need to have more cross reference to applicable parts of EU / PIC/S GMP in the revision of Annex 11 than exists currently. For example, Section 3 of the New Annex 11 is about pharmaceutical quality system (PQS) but does not mention Chapter 1.10
Naming convention: Clauses of the current Annex 11 will be A11 X and the new version as New X.Y. A similar approach is taken with Chapter 4: C4.X and New X.
Regulations: black, white or fifty shades of grey?
Regulations should be simple. The new draft has 111 clauses7 and is quite stringent and detailed trying to avoid interpretation by regulated users, whereas the current version is simple and needs interpretation.4 Linked closely with Annex 11 is the update to Chapter 4, which has ballooned from 32 to 85 clauses.7 These are paradigm changes.
Regulations should follow this approach:
- Regulations should define What has to be done.
- Each company interprets and decides How the regulations will be implemented.
- Regulators inspect to assess compliance with the regulations: How = What?
However, with the revision of Annex 11 and Chapter 4 there are many clauses that state How to comply rather than state What should be done.
Accelerate Mass Spectrometry Data Processing With Advanced Software Solutions
This case study reveals how advanced chromatography data system upgrades can deliver up to 15-fold increases in data processing speed, enabling comprehensive analysis of 250 components in under four hours instead of multiple days.View App Note / Case Study
Why are regulators trying to tell us how to work?
The revision often mandates how to comply; this regulatory overreach is illustrated in New 11.5:
… passwords should … contain a combination of uppercase, lowercase, numbers and symbols.7
This goes above and beyond what should be in a regulation and raises a question: how is this supposed to increase security?
Furthermore, password complexity is NOT a best IT practice. National Institute of Standards and Technology (NIST) Special Publication (SP) 800-63B (2017)11 and the July 2025 update12 discuss passwords in detail:
- Appendix A Strengths of Passwords: Reviewing password complexity: analyses of breached password databases reveal that the benefit of such rules is less significant than initially thought, and the impacts on usability and memorability are severe.12
- Section 3.1.1.1:
o Passwords SHALL either be chosen by the subscriber (user).
- Section 3.1.1.2:
o 1. … passwords that are used as a single-factor authentication mechanism to be a minimum of 15 characters in length.
A user should be able to select their own password.
o 5. … SHALL NOT impose other composition rules (e.g., requiring
mixtures of different character types) for passwords.
o 6. … SHALL NOT require … to change passwords periodically.
However, verifiers SHALL force a change if there is evidence that the authenticator has been compromised.
o The entire password SHALL be subject to comparison, not substrings or words that might be contained therein (against a block list).12
See also the article on passwords,13 which discusses the 2017 version of NIST SP800-63B.
Utah Medical vs FDA court case

Streamline Laboratory Software Management With Automated Updates
This case study demonstrates how one organization reduced update time from 3–5 days to just 30 minutes for 200 computers while eliminating travel costs and minimizing downtime.View App Note / Case Study
This is an example of a regulator telling a company how to qualify equipment and validate software. In 2005, FDA (Food and Drug Administration) versus Utah Medical court case centred on the company’s approach was different to FDA’s required approach. The judge stated: Many roads lead to Rome.14 FDA asserted that Utah Medical should comply with the 21 CFR 820 regulations in a manner satisfactory to the FDA. The judge stated that regulations are:
- … general and flexible so as to cover a broad spectrum of products and activities, …
Similar to the A11. - … have the virtue of generality and the vice of imprecision.14
Not black or white but fifty shades of grey depending on circumstances of an individual company such as size and the products being manufactured: solid dosage forms or advanced therapy medicinal products (ATMP). This is risk management in practice, as one size rarely fits all. - The judge found for Utah Medical as the company was in control of their equipment and software, even though they did not follow what was mandated by FDA.14 For further details, see the article by Burgess and McDowall.15
A lesson learnt?
Is there a lesson that can be learned from the update of Annex 1 (Manufacture of Sterile Medicinal Products)?16 This update, which came into effect before Annex 11, also ballooned in size to 59 pages and contains too much detail. Although quality risk management (QRM) is incorporated at the beginning into the scope, it does not provide the opportunity for independent risk-based implementation, which has led to varied interpretation of “required” language by global health authorities and by global manufacturers. In practice, some global regulators are not amenable to risk-based approaches (e.g., bracketing).
Annex 11 revision looks as if it is going the same way. Stakeholder feedback will be crucial here.
Concept paper on the revision of Annex 11
The concept paper on the revision of Annex 116 contained 33 topics for consideration when updating the regulation and requested industry feedback. Table 1 maps the areas in the concept paper against the clauses in New Annex 11.
Two key omissions are:
- Digitalization and technical controls over manual processes. Given the move to digital transformation under Pharma 4.0,17 this is a missed opportunity. This omission is compounded by 21 references to hybrid systems across the two updates.
- Cloud service providers (CSP) are not given the visibility they deserve, in contrast with the FDA guidance on electronic systems, electronic records and electronic signatures in clinical investigations: questions and answers,18 EMA guideline on computerised systems and electronic data in clinical trials19 and OECD GLP (good laboratory practice) 17 supplement 1 on GLP & Cloud Computing.20

Ask Me Anything: Next-Generation Battery Technologies
Join Dr. Boryann Liaw for a live Ask Me Anything on battery innovation. Explore solid-state modeling, fast charging, diagnostics and simulation. Gain expert insights into the future of electrification, renewables and sustainable energy storage.View Ask Me Anything
Table 1: Comparison of 2022 Concept Paper with New Annex 11 2025
| Clause | Subject | Annex 11 Clause(s)/Section(s) |
| 1 | Incorporation of the EMA Annex 11 Q&A | N/A |
| 2 | Data in motion / data at rest a | (10.1, 10.2, 10.3) 16 and 17 |
| Configuration hardening / technical controls | Not discussed | |
| 3 | Digitalization | Not discussed |
| 4 | No increase in risk when replacing a system | 2.8 |
| 5 | Reference to ICH Q9(R1) | 4.2 |
| 6 | Cloud service providers | Not discussed |
| 7 | Service provider agreements | 7.5 |
| Documentation available for inspections | 7.4 and 7.5 | |
| 8 | Commercial off the shelf (COTS) software | 6.1 (partially) |
| 9 | Defining qualification and validation | Glossary (poor) |
| 10 | Focus on testing GMP functionality | 9.6 |
| 11 | User requirements and traceability | 6.1, 6.4, 6.5, 9.5 |
| 12 | Agile software development | Not discussed (6.1?) |
| 13 | Definition of critical systems and critical datab | Not discussed |
| 14 | Protect GMP systems, networks and infrastructure and data | 15.1–15.20 and 16.1–16.5 |
| 15 | Testing data / archived data restores | 16.6, 17.4 and 14.2 |
| 16 | Backup expectations | 16.1– 16.5 |
| 17 | Electronic copies of data | 12.9 |
| 18–24 | Audit trails and alarms | 12.1, 12.2,12.3, 12.7, 12.8 and 8 |
| 25 | Configuration review | 14.1 and (6.6 partially) |
| 26 | System and data confidentiality, integrity and availability | 15.1 |
| 27 | Additional security controls | 15.1–15.20 |
| 28–30 | User authentication and access privileges | 11.2, 11.3, 11.10 |
| 31 | Validated archive processc | 17.1–17.5 |
| 32 | AI and ML(machine learning)d | Not discussed |
| 33 | Consider input from FDA draft computer software assurance (CSA) Guidance | Not discussed |
a For disposal of records see chapter 4.79
b Incorporated in chapter 4.13
c Also considered in chapter 4.76
d See Annex 22 (out of scope of this article)
Gone, but not forgotten?
In New Annex 11, existing key requirements from the current version have been omitted:
- A11 Principle: … IT infrastructure should be qualified.
This is an abysmal omission as questions have been raised already: as this has been omitted from the draft, given the prescriptive nature of the document, I do not need to qualify my infrastructure anymore? This is equivalent to building a house without foundations; see our comments under section 2. - A11 3.4 Audit information of suppliers
Availability of supplier quality system and audit information should be made available to inspectors on request is no longer in New Annex 11. - A11 4.3 Inventory and system description
The inventory of systems and system description for critical systems are omitted – why? These are critical omissions, as you need an inventory of systems for asset management as well as regulatory compliance. After a system risk assessment, ALL computerized systems will be listed in an up-to-date inventory so that items that are non-GMP equipment, instruments or systems in a GMP-area are properly identified and labelled Not for GMP use. - A11 4.7 Automated test tools
… Automated testing tools and test environments should have documented assessments for their adequacy. Given the rise of validation management tools since 2011, this paragraph should have been expanded and enhanced, not dropped. It also provides justification for not validating automated test tools, as some suppliers state these must be validated, but documenting them for adequacy.
o There is a mention of using a tool for traceability in New 6.5 but it is insufficient given the current availability of lifecycle tools.
o However, out of nowhere in New 4.23 and 4.25 there are mentions of automatic validation scripts,7 these are not equivalent to the use of automated test tools.
o There must be a section discussing regulatory requirements about tools supporting GxP/GMP activities such as automated testing tools and validation management systems but it must be in Annex 11 not in Chapter 4.
- A11 4.8 Data migration
If data are transferred to another data format or system, validation should include checks that data are not altered in value and/or meaning during this migration process.4
New 10.3 has a clause on data migration, but does not cover the alteration in value and/or meaning. - A11 8 Printouts
These are no longer required during an inspection, the emphasis is on e-records. A change for the good. - A11 13 Incident management
IT incidents and problems are key inputs for periodic reviews. Again, another critical omission from the revision. - A11 15 Batch release
Release of batches by a qualified person (QP) using an electronic signature is omitted but this is covered in Annex 16.21 In New Annex 11, it is replaced by a requirement to allow a QP access to audit trail entries. Good luck with that! A QP should always have access to all data and information relevant to the batch, where necessary they perform a review with the help of expert(s). - Glossary
Process and system owners are deleted: this avoids allocation of responsibilities. Furthermore, IT incidents and problems are not defined nor are IT changes. Glossaries are discussed later in this article.
Regulatory plagiarism?
It is déjà vu all over again with some of the clauses in both Annex 11 and Chapter 4. Where have they come from? See Figure 2.
- An information security management system (ISMS) under ISO 27001 turns up again in New 15.1. An upper-case ISMS was proposed in the 2008 update of Annex 113 and rejected by the industry but makes an unwelcome lower-case return.7 Second time lucky? Mentioning this implies that an ISO 27001 ISMS22 should be implemented and certified, which may be impossible for small regulated users.
- PIC/S PI-04123 provides input to many sections to Chapter 4. This is not surprising as section 3.7 of PIC/S PI-041 states This guide is not mandatory or enforceable under law.23 What you see is a distillation of key parts of PIC/S PI-041 guidance into regulation, e.g., control of blank forms is in guidance documents and is incorporated in Chapter 4 to become a regulation.7 Ideally, you need to digitalize processes to get rid of paper.
- WHO’s TRS 1033 Annex 4 DI guidance from 202124 provides some input to New Chapter 4.
- The biggest copy and paste source is from a GLP document: OECD GLP 25 on IT security,25 published in late 2024, which can be mapped completely or in part to 10 clauses in Annex 11 Section 15 and 5 clauses of Section 16, which are copied either verbatim or with minor modifications (see Table 2 for the example for anti-virus software). Considering the different context within GxP, the question is whether it is beneficial to push and integrate GLP requirements and definitions into GMP? The answer is outside of our discussion in this article.
- OECD GLP 17 on Computerised Systems is the verbatim source for nine of the entries in the New Annex 11 glossary.26 A note to the authors, copy and paste is great but the definition for COTS software still contains the GLP term test facility management (TFM). Doh!
The problem with COTS is that the term is not consistent with GAMP 5 SE.27 We will discuss COTS later in this article.
Figure 2: Input sources to the proposed revision of Annex 11 and Chapter 4. Credit: Bob McDowall.

Cloud-Based Digitalization Solutions Powering the Lab of the Future
This whitepaper explores how labs can embrace digitalization for better connectivity and future-proofing processes against growing GxP requirements.View Whitepaper
Table 2: Sections on anti-virus software from OECD GLP No 25 and New Annex 11
| OECD GLP 25 | New Annex 11 |
| 9 Anti-virus software should be installed and activated on systems used in GLP, as appropriate. The anti-virus software should be continuously updated with the most recent virus definitions in order to identify, quarantine, and remove known computer viruses. This process should be monitored. | 15.18 Anti-virus software should be installed and activated on systems used in GMP activities, especially those interfacing the internet. The anti-virus software should be continuously updated with the most recent virus definitions to identify, quarantine, and remove known computer viruses. The effectiveness of the process should be monitored |
Note: words not matching are in italic font and underlined.
Section 2 principle
As mentioned earlier, the following ten words must be retained in this section:
The application should be validated; IT infrastructure should be qualified.
This clearly encapsulates the essence and one of the key requirements of Annex 11. Omission in the released version would be an abrogation of duty by EMA and PIC/S.
- 2.2 Quality risk management
Section 4 is on QRM and refers to ICH Q9 (R1).28 QRM is repeated in New 4.3, 6.2, 7.2 and 9.2, which agree that the level of efforts for system validation should be defined based on the system risk assessment and the intended use. QRM is parroted throughout both revisions when all that is required is a single mention. - 2.4. Data integrity
DI is discussed in sections 10, 11, 12 and 13. The expansion is to Security and data retention.
New 2.4 mentions ALCOA+, while the proposed revision of Annex 11 lacks the discussion of this principle. There is also a bad day at the coordination office as Annex 11 mentions ALCOA+ and New 4.63 ALCOA++ with the emphasis on traceability, similar to the update to USP <1029> on Good Documentation Guidelines and Data Integrity.29 This disconnect is carried through to the Glossaries in both documents, as we critically review later. - 2.6. Outsourced activities
Section 7 explains the qualification requirements for vendor, service provider and internal IT, however it lacks any clause on cloud services. Use of cloud was a reason for the Annex 11 update, as stated in the concept paper and the New Annex 11.6,7 - 2.8. No risk increase
The statement in the New Annex 11, Where a computerised system replaces a manual operation, … There should be no increase in the overall risk of the process, is extended to include replacement of one system by another. This is the second key requirement of Annex 11 since 1992. Why has it been relegated to the back section of the principle, instead of keeping it as a main principle?
The principle can be condensed greatly because much of the content is repeated later in the document.
Section 3 pharmaceutical quality system (PQS)
Why repeat what is already in Chapter 1 PQS10 and Chapter 9 self-inspections30 in clauses 3.1 i and 3.1 iii? Why create repetition and redundancy? Cross reference would be better.
Clause 3.1 ii covers changes to a computerized system, however A11 10 has a separate section and heading which highlights the importance of Change and Configuration Management which is hidden in the update.
A good point is the reinforcement of the role of management in the implementation and operation of computerized systems in New 3.1 iv and v.
BUT, there is no point having management in charge of computerized systems if they don’t understand them. Tender Corporation and Stason Pharma were cited in FDA warning letters31,32 for lack of senior management and quality assurance oversight of computerized systems, for further reading see the analysis of these two warning letters.33
We suggest incorporating these in section 5 personnel and training but reverse the order of these two sections to enhance the responsibilities of management and their training.
Section 4 risk management
A11 1 on risk management is very simple: apply risk management throughout the lifecycle.4 This can be interpreted as necessary depending on the risk posed by the system and the required controls to ensure the integrity of the data.

Tech Spotlight: Lab of the Future 2025
Discover how innovations in lab informatics, robotics and real-time data integration are driving enhanced research efficiency, fostering collaboration, and ensuring data integrity.View Webinar
In section 4, we have five clauses going into detail on lifecycle identification and analysis, appropriate validation, mitigation and DI. In addition, 4.5 of New Annex 11 discussed data vulnerability under DI. Our suggestion is to rephrase the subtitle into Data risk to stay consistent with New Chapter 4 (4.13) and PIC/S PI-041 section 5.5.23 Alternatively, record risk is a better phrase. As discussed earlier, QRM or risk management is referred to ten times in New Annex 11, which is too much and too detailed.
Take A11 clause 14 and add business capability or process understanding as the fourth risk criterion and reference to ICH Q9(R1)28 is all that is required.
Section 5 personnel and training
New 5.2 mentioned … adequate system specific training … Along with the GMP awareness, training on data security principles is a requirement as stated in New 15.3 and is consistent with section 5.3.2 of OECD GLP 17 supplement 1.20
However, there is no mention of any assessment of competence or understanding. Competence needs to be ensured by different choices, e.g., using a questionnaire34 or simulated tests for cybersecurity (New 15.3). Again, the latter option in New 15.3 centred on How to do, not What to be achieved.
To find any training assessment, you need to go to New 4.45 (vi) … verification of the effectiveness of training. However, EU / PIC/S GMP Chapter 2 clause 2.11 on training states that its practical effectiveness should be periodically assessed.35
The two clauses on the role of management should be placed here, as mentioned in Section 3 of this article. Don’t forget that management need training too, especially on the implementation and validation of computerized systems!
Section 6 system requirements
A11 4.4 simply states:
User Requirements Specifications (URS) should describe the required functions of the computerised system and be based on documented risk assessment and GMP impact. User requirements should be traceable throughout the life-cycle.4
A simple and easy to understand clause: you need a URS and any further specification documents to adequately define the intended use of the system. A URS is the most important document in any validation project as it defines the intended use; no URS, no validation. Requirements should be traceable – the means of how to do this is left open.
However, both the existing and New section 6 do not state intended use coupled with underlying process understanding and, where appropriate, improvement.
New Annex 11 has 6 clauses to describe how to document user requirements, application configuration and mandates a traceability matrix. All you need is a way to demonstrate traceability – how this is achieved is up to a regulated user. Traceability from the URS should also include SOPs, supplier assessment, installation and commissioning by a supplier as well as a validation summary report (VSR).
A good VSR is vital as it is the starting point for regulatory inspections as per clause 23.10 of PIC/S PI-011.36 BUT a VSR is not defined in GMP regulations. Section 9 of New Annex 11 is deficient as there is no formal mechanism for system release discussing the overall validation – warts and all – by not concealing any problems encountered in a project. Therefore, a VSR should be defined in Annex 11.
A good point that has been added is that user requirements are living documents and must be updated but version controlled (New 6.4). As knowledge of the system develops or more functions are used, the URS and other specifications must be updated, with consequential knock on to other validation documents as necessary, e.g., test scripts.
Configuration of an application is important and must be prospectively specified and documented in a separate configuration specification (New 6.6), depending on system complexity. However, the problem with New Annex 11 is that configuration can refer to two areas:
- Application configuration: the technical control settings to ensure DI and data security, e.g., definition of user roles with access privileges.
- System configuration: a list of all components of a system, such as virtual or physical computer system, data storage, peripherals, operating system, utilities, middleware, database and application.
Both are essential for change control. However, the Glossary of the New Annex 11 only refers to system configuration under the term configuration.7

Innovations in Cell Line Development
This eBook explores strategies, innovations and integrated solutions to streamline every stage of cell line development.View eBook
Section 7 supplier and service management
This is expanded from A11 3 and now consists of 5 clauses. How much overlap is there with Chapter 7 on outsourcing? 9 A good point in New 7.1 is that the responsibility and accountability remains with the regulated user of service suppliers, including the company’s IT Department. New 9.9 reiterates this point under Authorisation.
Supplier assessment is now an audit or a thorough assessment in New 7.2, rather than a need for an audit should be based on a risk assessment. It appears that assessment questionnaires are consigned to the round filing cabinet of history as useless? See the article on selection of a CSP37 to separate clouds from clods.
Requirements for contracts are very detailed in New 7.5 and appears as a “how-to write a contract” instruction. There is not a specific or detailed section on cloud computing which is only mentioned in passing.
There is a disconnect in New section 7 regarding the requirement on service level agreements (SLA):
· New 7.3 describes the level of oversight through defining SLA and key performance indicators (KPI) for service provider or an internal IT department,
· New 7.5 mentions … the regulated user should have a contract with a service provider or have approved procedures with an internal IT department.
New 7.5 (Contracts) – should this level of detail be in a regulation? If it remains, the clause should be re-ordered for better understanding: responsibility, audit, contracts, documentation availability and oversight. For more detail on the scope, including use of subcontractors by a supplier and content of SLA, see our complete article.38
There is nothing in detail on the cloud and the role of a SaaS (Software as a Service) supplier in aiding inspections. Better sources of regulatory guidance are:
- Section C on information technology service providers and services sections of the FDA guidance for electronic systems, electronic records, and electronic signatures in clinical investigations. 18 This guidance also provides simpler and easier to read sections on both application and IT services compliance.
- Section 6.7 and Annex 1 on cloud solutions and agreements in the EMA guideline on computerised systems and electronic data in clinical trials. 19
- OECD GLP 17 Supplement 1 on GLP and cloud.20
Perhaps back to the future is called for by retaining IT-departments should be considered analogous in the current 3.1 and equating it to an external service provider?
Section 9 qualification and validation
How about this for another bad day at the office? Annex 11 says follow Annex 15 for qualification and validation and Annex 158 says follow Annex 11 for validation of computerized systems, as shown in Table 3. A regulatory Groundhog Day? Who has not read Annex 15?

Master Seamless CE-SDS Method Transfer
This whitepaper presents validated bridging protocols and real-world case studies demonstrating successful method transfer strategies.View Whitepaper
Table 3: Cross-reference confusion between Annexes 11 and 15
| New Annex 11 | Current Annex 15 |
| 9.1 Principles. Qualification and validation activities for computerised systems should follow the general principles outlined in GMP Annex 15. | Principle: Computerised systems used for the manufacture of medicinal products should also be validated according to the requirements of Annex 11. |
Even if you follow the qualification and validation section in Annex 15, we meet the 4Qs model which should have been pensioned off long ago. FDA discontinued the use in 2002 with the general principles of software validation39 and GAMP 5 stopped using 4Qs in 2008.40 Also Annex 15 mentions factory acceptance tests (FAT) and site acceptance tests (SAT) which are typically applicable to production systems.8
The separation of specifications in New section 6 from the remainder of the lifecycle is strange: sections 6 and 9 should be integrated together, as it is in the current Annex 11, and come after section 7, e.g., assess supplier(s), specify and validate the system.
New 9.4 test evidence should follow GAMP 5 SE, stating primary test evidence and secondary test evidence: use the audit trail to document testing and only use screen dumps when necessary, e.g., to trace the actions back to the requirements.27
This section only considers the execution of test scripts. However, critical requirements are also verified via writing SOPs for system use, execution of supplier installation and commissioning (installation qualification (IQ)/ operational qualification (OQ)) protocols, which are not covered. Written instructions (e.g., SOP) are also required to perform the validation. More details on Procedures can be found in New 4.27.
New 9.7 Validation plan should be established after system selection and the impact/risk assessment. It is formal evidence of validation that is in the scope of audit and inspection. Our suggestion is to discuss it under 9.4.
CSA is dead as a dodo!
FDA draft CSA guidance41 allowed the use of unscripted testing for medical device quality system and production software. As a result, many have jumped on the bandwagon of unscripted testing to be applied to Pharma.
New 9.4 and 9.7 state:
System qualification and validation should provide evidence in the form of executed test scripts …
… Test scripts should be described in sufficient detail to ensure a correct and repeatable conduct of test steps and prerequisites.
The requirement for test scripts to be described in sufficient detail for … repeatable conduct is a complete rejection of CSA unscripted testing.
However, there should be emphasis on using trained users to write and execute test scripts.
How much detail is adequate? This is in the eye of the inspector but commensurate with the system risk.

Modernization of Liquid Chromatography Methods
This webinar features two expert speakers who will share insights into the latest LC methods and technologies.View Webinar
A test script must contain sufficient detail to execute instructions along with acceptance criteria that are clear and concise. In a laboratory, a trained analyst knows what to do when given an instruction in an analytical procedure: prepare 1L 0.1M sodium acetate solution. This involves:
- Calculating the weight of sodium acetate necessary
- Checking the analytical balance is qualified and works within limits
- Weighing the material
- Transferring it to a volumetric flask
- Dissolving the solid and making up to volume
- Labeling the storage vessel with contents, name of analyst and dates or preparation and expiry
- All work being recorded as necessary with material name, expiry date and batch number, balance printout, etc.
Similarly, a test script should not require every mouse click or menu option to be specified and tested. Trained users are not idiots; regulations should not treat them as such.
Section 10 data handling
Some of the new requirements are similar to existing A11 clauses.
- New 10.1 verification of manual data entry equates to A11 6 (Accuracy Checks)
- New 10.2 (Data transfer) equates to A11 5 (Data).
Data migration is discussed twice in New Annex 11:
- New 10.3 indicates … Where an ad hoc process requires that critical data or a whole database be migrated from one system to another …
- New 15.11 states Validation of applications on updated operating systems and platforms and migration of data should be planned and completed in due time prior to the expiry of the vendor’s support.
From experience, data migration is not an ad hoc process. NEVER undertake an ad hoc migration; data migration needs to be carefully planned. The source and destination systems and the way each one stores data must be fully understood. A case study of data migration can be found in the book on validation of chromatography data systems (CDS). 42 C4.10 and New 4.76 both mention controls are required to ensure the integrity of the record throughout the lifecycle.5,7,43
Section 11 identity and access management
From 10 words in A11 12.3, we now have another example of regulatory bloat to 11 clauses and prescription of what to do. Apart from the detailed requirement in New 11.5 (secure passwords) that was discussed earlier in this article, we have the following:
- New 11.1 states … The use of shared accounts except for those limited to read-only access (no data or settings can be changed), constitute a violation of data integrity.
The use of shared read-only accounts is a poor IT practice. If found in an audit, there is always a question: are there more serious examples of account sharing? - New 11.6 provides another example of saying how to do instead of what to be done. Multifactor authentication (MFA) is one of the possible technical options (today) but may not be useful in the future.
- New 11.8 on inactivity logout is in line with PIC/S PI-041 9.5. no 123 Systems should include an automatic inactivity logout, which logs out a user after a defined period of inactivity …
An automated inactivity screen lock can present a major problem in an analytical laboratory. Many laboratory systems are standalone and will run unattended overnight. If an inactivity logout is used, systems may stop operating unless there is a functionality to permit a user to log out and this allows the sequence to still operate.
In this section, there are also good requirements for:
- Unique accounts
- Confidential passwords
- Segregation of duties
- Least privilege principle
- Checks to ensure that user accounts are still required
However, you should have been doing this already!
One point that is missed in this section is about the system’s ability to generate a list of users and their user roles / access privileges as discussed in PIC/S PI-041 9.5. 23 This is a helpful requirement that should usually be addressed in a URS.

Advanced Solutions for Mining and Metallurgy
This guide highlights a complete portfolio of spectroscopy, chromatography and mass spectrometry solutions that support efficient analysis at each step, from exploration to refining.View How To Guide
Section 12 audit trail (AT)
There are two updates, which are positive:
- Reviews. It is essential to have an SOP for each system AT review. This is consistent with our recent AT article44 but is contrary to PIC/S PI-041, section 9.8 no 123 that only requires a single AT review SOP.
- Electronic copy. Electronic AT entries are now required instead of a printout.
However, there are poorly written clauses:
- The first clause on user interactions is inadequate, as an AT must capture system actions as well. Imagine if you have an overnight run in a laboratory: You start the sequence, which is logged in an AT. You go home and the rest of the work is captured automatically; no manual involvement, so no AT entries?
Both operator interaction and system entries are generating GMP data and as the glossary states, AT allows reconstruction of the events so we should have a similar AT functionality for system and user interactions.44 - What about automatic data transfer? There should be an entry in the source system AT (e.g., CDS) and a corresponding one in the destination system AT (e.g., laboratory information management system).
- New 12.3 No edit or deactivation: AT must be configured and activated at installation and cannot be turned off (good). Concept paper item 18 states AT functionality is mandatory and any grace period has long expired,6 but New section 12 fails to mention it explicitly.
o The ability to deactivate an AT by a system administrator is totally unacceptable and must be deleted from this regulation.
- New 12.4 lacks discussion of review by exception mentioned in section 9.6 no 1 of PIC/S-041.23
- An option to Export of the data to a tool is slow and if the export is text, it increases the risk of DI violation.
- The clause on Independent review needs clarification that the originating department must perform AT review as stated by PIC/S PI-041 section 9.6 no 123 and the FDA DI guide, Q7.45 In addition, Q16 asks Should personnel be trained in preventing and detecting data integrity issues as part of a routine CGMP training program? Yes.45
- New 12.7 omits the criticality of data and metadata assessment that need to be reviewed.
- Availability to QP. Good luck with this one! Probably, a QP will need help from a subject matter expert (SME) as most likely they are not a user of the system. What about the other e-records on a system, are they off limits?
Section 13 Electronic signatures
An earlier article by one of us compared the e-signature requirements for A11 14 with those in 21 CFR 11 and questioned why the FDA required 640 words (not including definitions) to achieve what Annex 11 achieved in 40 words?46 This was the power of interpreting the phrase electronic signatures … have the same impact as hand-written signatures within the boundaries of the company.4
The proposed version now has 9 clauses and 357 words. Only another 283 words to catch up with the FDA!
This section must be read in conjunction with clauses New 4.64–4.75 on signatures in GMP relevant documents.
New 13.6 Manifestation: there are differences between New Annex 11, 21 CFR 11 and even New Chapter 4 (see Table 4).
Table 4: Comparison of electronic signature requirements of 21 CFR 11, New Annex 11 and New Chapter 4

The Science That Keeps Biofuels Flowing
This infographic explores how to apply the right analytical technique during multiple stages of bioethanol and biodiesel production.View Infographic
| New Annex 11 (13.6) | 21 CFR 11.50(a) | New Chapter 4 |
|
|
|
The additions of user identity and the role of the signer add zero value to an electronic signature. New 4.73 agrees that the meaning of an e-signature implies the role of the signer.
Further comparison is about New 13.4 and New 4.65, indicating the use of date and time. The question is if date and time is mandatory with a signature on a paper GMP record? For an electronic signature, yes, it is, because it automatically timed and dated, however for handwritten signature it would be date only. Why sign a record with time on paper? This is an over burdensome and unnecessary requirement.
New 13.8 Unbreakable link Electronic signatures should be permanently linked to their respective records. Controls should be in place to ensure that a signed record cannot be modified or alternatively, that if a later change is made to a signed record, it will clearly appear as unsigned.
- The first sentence is fine, but the underlying e-record set must be locked and cannot be changed.
- What 21 CFR 1147 or New 13.8 both omit is the issue of revoking an e-signature. In the cellulose world, if an approved report is found to have a mistake, the report is recalled, the mistake corrected, approved and reissued. The document history will record the update and the original report will still be available as an archived version.
- If a signature has been removed and the record is now unsigned could this be used as a means for falsifying data?
- Removing an e-signature from an electronic record compromises ALCOA++ criteria. The e-signature removal means that a record is not complete, not accurate and work is certainly not traceable.
- In an electronic world, you need a function to revoke electronic signature(s); the signatures still be visible on the report, the reason for revoking must be documented on the record and in the AT, e.g., a mistake should be explained, such as a miscalculation. When corrected, the report is re-signed electronically and reissued. The whole signing sequence: original e-signatures, revoking e-signatures and e-resigning must all be available to ensure traceability (the tenth ALCOA++ criterion – but obviously only in New Chapter 4).
New 13.9 will be discussed later under hybrid systems.
Signatures in GMP documentation: attribution versus signing
Nearly 30 years ago, following the publication of 21 CFR 11 for electronic records and electronic signatures, 47 we had a debate and resolved the difference between attribution of action versus signing of a record in a computerized system. There are very few requirements for signing records: reporting final test results, certificates of analysis, qualification and validation documents, method validation protocols and reports, etc. However, sections on signatures in New 4.64–4.75 and Section 13 in New Annex 11 reopen that debate, as there is no stated separation between attribution and signing, only the requirement for signatures.
It appears at first sight everything must be signed, but finally in New 4.68 the regulated user should have identified those records which require a legally binding signature. This should be the first requirement in this section.
New 4.64 Signatures are essential for ensuring accountability …
Does every action on paper or using a computerized system now require a signature? No, identification of an individual performing an action is the key objective.
There are many superfluous requirements in clauses 64–65 of New Chapter 4.
Use of initials, common in some countries, requires a procedure to permit this. In EU GMP Chapter 6 (Quality Control) there are only requirements for initials for testers and reviewers in clause 6.17 (f) and (g) and the only signature requirement is for the batch release in clause (h).48
Section 14 periodic review (PR)
The update is more detailed which is good; covering not just ensuring the validation status of the system but also the use of it, e.g., check AT review effectiveness. New 14.1 requires a report containing findings and corrective actions, which will be checked in the next PR of the system as stated in New 14.2 iv.
New 14.3 introduces risk-based frequency of PR considering system criticality and data risk and vulnerability so the frequency of PR will vary between systems. This is important as many organizations push a PR for all systems to three years – this is lazy and wrong, as some systems are higher risk than others. We suggest that for critical systems, a PR six months after implementation should be good practice to ensure all is working correctly, as waiting two to three years would be too late to correct any problems. The issue of frequent updates under SaaS agreement is not addressed in this section. For practical information and advice on PR, see.42

Analytical Action To Ensure Water Availability in Semiconductor Manufacturing
This whitepaper examines innovations in water usage and recycling within the semiconductor industry, exploring the analytical challenges in elemental contamination monitoring.View Whitepaper
Section 15 security
Oh dear, this is a shopping list of 20 clauses that is brutally prescriptive and should have no place in a regulation. This raises the question of verification by inspectors, who normally have no expertise in this area. As noted in Figure 2, ten sections have been lovingly ripped off from OECD GLP 25.25 These 20 tasks are what any competent IT department or service provider should be undertaking already.
New 15.2 Continuous should be continual to be consistent with GMP Chapter 1.10 Continual is a discontinuous process that allows for change control, which is critical for ensuring compliance. You may be assessing cybersecurity threats continuously but updating systems continually.
Disaster recovery (DR) plan is described in New 15.7; however, it is insufficient in addressing DR elements:
- Both recovery point objective (RPO) (how much data can you afford to lose) and recovery time objective (RTO) (the time it takes you to recover the data or system) must be defined in a DR plan.
Why does New 15.7 only consider RTO? - Business continuity (BC) is centred on alternative working arrangements. 27 However, with increasing digitalization of Pharma, alternative working practices based on paper are insufficient, therefore maintaining computerized system operations is critical and requires failover in separate locations (see New 15.6 Replication).
- DR and back up (section 16) are interconnected: without back up there is no DR.
- New 15.15–15.17 allow the use of USB (universal serial bus) devices. These have major security and data vulnerability issues and should be avoided totally.49
Section 16 backup
Backup is expanded from one to six clauses in the new version based on OECD GLP 2525: much of this is common sense and begs the question, why should it be in a regulation? If this level of detail is required, then why not add a clause requiring a backup of data, application and system before making a change? In case of problems, you know it makes sense.
Replication is also discussed in New 15.6 but this is part of an overall backup strategy, given the dependency of Pharma on computerized systems.
A11 7.2 states … Integrity and accuracy of back-up data and the ability to restore the data should be checked during validation and monitored periodically. The clarity and simplicity of this sentence is lost in New 16.6 as it implicitly mentions … tested and documented based on risk during system validation …
The content in New 16.1–16.4 is also repeated in New 4.76.
Section 17 archiving
The following combines our comments on section 17 and New 4.76–4.79 for electronic data.
Rather than archive laboratory data off-line, it may be better to archive in the system that created the records. This has the advantage that both archived and live data can be updated if necessary and avoids the need to check the readability of archived data. However, this will depend on the storage available, adequate back up with fail-over and if functions in an application permit archived data to be locked and kept secure. See OECD GLP 15 section 8 for a better discussion of an electronic archive.50
An alternative viewpoint is NIST SP800-209 for Security Considerations for Storage Infrastructure, which suggests:
IS-SS-R1 (b) Long-term archive and backup systems should be separated from production data storage systems.51

Detection of Endogenous Biomarkers in Complex Matrices
This webinar presents a highly sensitive LC-MS/MS workflow that overcomes the limitations of traditional methods and allows for the detection of biomarkers in keratinized matrices.View Webinar
There may be archive on the production system but it is imperative that there are recovery copies on separate off-line and off-site storage. Regardless of where data are stored, a regulated user must ensure long-term availability of data.
One issue that is not mentioned in New 4.79 on avoid destroying the wrong records at the end of the data lifecycle, is if there is any legal action pending? Whilst not a regulatory issue, it is a legal one and some records may require a legal hold to prevent destruction even if the retention period has expired.
New 4.26 reiterates that dynamic data should not be converted to static data; this is a good point and is consistent with existing guidance documents.23,45,52
Retention of documents: definition of location
The potential problem with both New Annex 11 (16.4) and Chapter 4 (4.76) is that there is no definition of location, which is similar in GLP regulations.49,53,54 Location of paper records is straightforward either in a building on site or in a document repository: location is the street address. However, what is the situation if using a CSP? Will a uniform resource locator (URL) be adequate or do you need the address of the data centre? This was the question posed in our recent article on SLA38 and also in section 6.3 of OECD GLP 17 supplement 1.20
Many internet service providers (ISPs) will not provide the address of their data centre as this is a security breach. Regulators must bring their regulations into the real world and decide if a URL is an adequate location for access to a cloud repository.
Chapter 4 documentation and its impact on Annex 11
The current Chapter 4 is relatively concise with a principle and 32 clauses. It has expanded greatly in the revision to a principle and 85 clauses. This chapter is devoted to documentation, however if the GMP documentation is kept as a set of electronic records, the requirements of Annex 11 must be applied and there is no need to discuss electronic matter again in Chapter 4. Why repeat Annex 11 requirements in Chapter 4? This causes confusion and makes effective interpretation (where allowed) much more difficult.
Principle: a critical missing word
The Principle of the current Chapter 4 begins with Good documentation, to set expectations for the reader. 5 New Chapter 4 goes downhill immediately by just stating Documentation. Readers must wait until New 4.56 for a mention of Good documentation.7 Why delete a critical word at the very start of Chapter 4 and set a low expectation compared with the current version?
New 4.8 makes clear that all documentation, regardless of creation in-house or outsourced, must meet GMP requirements for legibility, accuracy, integrity and completeness.
Hybrid systems in Chapter 4?
Hybrid systems were not discussed in the concept paper for Annex 11.
6
The 2016 WHO TRS 996 Annex 5 contains the phrases

Expand NPS Detection With High-Resolution Screening
This app note highlights a LC-MS/MS workflow that enhances NPS detection, enabling confident screening of over 900 compounds in complex biological matrices with a single injection.View App Note / Case Study
- The use of hybrid systems is discouraged
- Replacement of hybrid systems should be a priority
The same approach is taken by PIC/S PI-041.23 To quote that eminent GxP expert, William Shakespeare: To hybrid or not to hybrid, that is the question!
Yet when we come to New Annex 11 and Chapter 4, hybrid systems are mentioned 19 times in New Chapter 4 and twice in New Annex 11. The closest we come to digitalization in the two updates is New 4.70:
If records exist electronically such records should be signed electronically. The use of a hybrid system should be avoided.
If signatures exist parallel in paper and electronically (e.g., in hybrid systems), the regulatory relevant signature should be defined by the regulated user.7
The first sentence is great, provided that the system has electronic signature capability and, more importantly, a regulated user implements, validates and uses it. The latter is typically the rate-limiting step; paper trumps e-records in many organizations. The problem is that there is more information in an electronic record than can be found with a printout. Q4 of the records and reports level 2 guidance on the FDA website,56 Q10 in the FDA DI guidance45 and OECD GLP 17 clause 10726 all mention electronic data is the true GMP record as they contain more information than paper printouts.
The four clauses for hybrid systems in New 4.82–4.85 require:
- Risk-based validation and control but hybrid systems present high DI risk anyway.
- A detailed description of the entire system. Why do hybrid systems only require a system description, especially when this has been dropped from Annex 11?
- The interface between manual and the computerized system must be managed and controlled with yet more QRM. This is simply restating the clause on accuracy checks in A11 6 using more opaque and wordy language.
- Evaluation, approval and archiving of data of both e-records and paper require procedures.
What is the point of these clauses when they simply restate what is already in Annex 11?
The signature requirement in New 13.9 for hybrid systems presents a technical challenge for suppliers and regulated users alike:
13.9. Hybrid solution. If a wet-ink signature … is used to sign electronic records held in a computerised system (a hybrid solution), measures should be implemented to provide a high degree of certainty that any change to the electronic record will invalidate the signature. This may be implemented by calculating a hash code (check sum) of the electronic record and printing that on the signature page.7
- What is this doing in a section on electronic signatures when it should be in Chapter 4 or a dustbin? It would be much better if the regulations persuaded regulated users to avoid using hybrid systems as advocated by WHO and PIC/S guidance documents.23,55
- It is inappropriate and a prescriptive regulatory overreach.
- Currently, it is unlikely that any hybrid system could meet this requirement.
- It is highly unlikely that suppliers would develop such a functionality.
- If implemented in the final release of Annex 11, all hybrid systems are out of compliance immediately.
Glossary problems, omissions and inconsistencies
There are problems with glossaries, not just with the New Annex 11 and Chapter 4,7 but also with A114 glossary and EU GMP glossary.57 Comparison of selected entries across all four glossaries is shown in Table 5.
More worrying are the omissions. For example, there is no definition of deviation in any EU GMP glossary.
Table 5: Annex 11, Chapter 4 and EU GMP Glossary consistencies and inconsistencies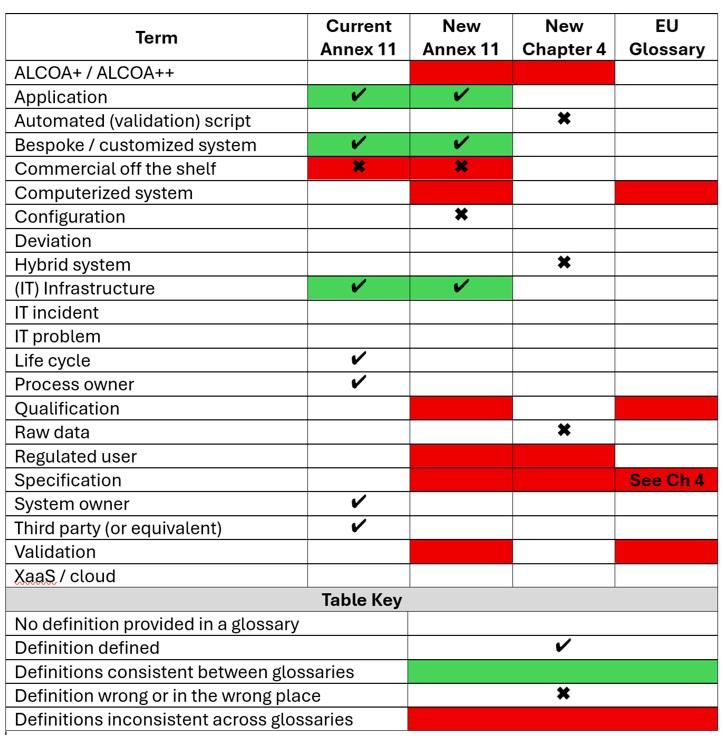
Glossary omissions
The following terms in A114 have been omitted from the revised version and need to be included in the final version of the update:
- Process owner
- System owner
- Third party
- Lifecycle: At least this needs to be defined on a general level covering computerized systems.

AI-Powered Lab Platforms: From Data Overload to Data Fluency
This whitepaper presents AI-powered SaaS, the next advancement in lab informatics that is transforming how lab workers interact with their data, enabling natural language conversations that deliver contextualized insights exactly when needed.View Whitepaper
Undefined terms
An IT incident or problem is usually not a deviation but many QA departments classify them as such. This is despite the wording in the current and proposed revision of Annex 11 in PR: incidents, problems and deviations are separate items.4,7 An IT incident, such as a backup failure, usually has no impact on product quality or patient safety and therefore should not be classified as a deviation. However, if an incident is not handled in a timely and adequate manner, this could lead to a deviation.
Any definition of cloud services has been omitted from the Annex 11 glossary. Given the use of cloud in Pharma this is a critical omission.
Raw data definition is wrong
Raw data is a GLP term that has been available since 1978 in 21 CFR 58.3(k):
Raw data means any … records … or exact copies thereof, that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study …53
The inclusion of raw data into EU GMP began in 2011 with the current version of Chapter 45 but with no definition. The MHRA 2018 GXP DI guidance defined raw data as:
Raw data is defined as the original record (data) which can be described as the first-capture of information …52
The explanation, but not the definition, then states Raw data must permit full reconstruction of the activities.52 However, only the MHRA definition was copied into PIC/S PI-041 as:
Raw data is defined as the original record (data) which can be described as the first capture of information, … 23
However, the reconstruction of the report has been lost. This error is then transferred to the updated Chapter 4 definition of raw data.7
A better approach would be to use the phrase complete data from 21 CFR 211.194(a).58 Further reading on this subject can be found here.59,60
Glossary inconsistencies

Lab Sustainability: From Paperless Workflows to Energy-Wise Labs
This eBook explores practical strategies to reduce environmental impact while maintaining research integrity.View eBook
Table 6: Glossary inconsistencies
| Term | Definition 1 | Definition 2 |
| ALCOA | ALCOA+ | ALCOA++ |
| Computerised System | A computerised system is a function (process or operation) integrated with a computer system and performed by trained personnel. The function is controlled by the computer system. The controlling computer system is comprised of hardware and software. The controlled function is comprised of equipment to be controlled and operating procedures performed by personnel. | A system including the input of data, electronic processing and the output of information to be used either for reporting or automatic control. EU Glossary57 |
| Qualification | Action of verifying that the system (including hardware and software) is effectively designed, installed, commissioned, and operates correctly. Refer to Computer System Validation New Annex 117 | Action of proving that any equipment works correctly and actually leads to the expected results. The word validation is sometimes widened to incorporate the concept of qualification |
| Regulated User | A company regulated under GMP | Marketing Authorisation Holder, Manufacturers, control laboratories, importers, and wholesale distributors (if the wholesale distributor holds a manufacturing license) |
| Specification | A document that specifies, in a complete, precise, verifiable manner, the requirements, design, behaviour, or other characteristics of a system or component, and often, the procedures for determining whether these provisions have been satisfied New Annex 117 | A list of tests, references to analytical procedures, and appropriate acceptance criteria that are numerical limits, ranges, or other criteria for the test described. It establishes the set of criteria to which a material should conform to be considered acceptable for its intended use. “Conformance to specification” means that the material, when tested according to the listed analytical procedures, will meet the listed acceptance criteria EU Glossary refers a reader to Chapter 4 for the definition57 |
| Data Risk Assessment | The process of evaluating the risks associated with the regulated user’s data. It ensures an efficient and effective approach to data integrity by considering the vulnerability of data to involuntary or deliberate alteration resulting in risk- based control measures. New Chapter 4 (Line 537-540)7 | The process of evaluating the risks associated with the regulated user’s data. New Chapter 4 (Line 589)7 |
New Annex 11 and Chapter 4 are inconsistent as the former uses ALCOA+ and the latter ALCOA++. USP <1029> update on good documentation guidelines and data integrity, issued a week before the New Annex 11 draft, uses ALCOA++ with heavy emphasis on traceability.29 More detail on its traceability is the glue for ALCOA criteria can be found here.61
The definition of Commercial off-the-shelf has been copied from OECD GLP 17: … if provided by a vendor to the general public, if available in multiple and identical copies, and if implemented by the test facility management without or with some customization.26 There are three problems:
- As mentioned before, the definition retains the GLP term of test facility management.
- Commercial must be removed, as GAMP 5 SE has renamed category 3 software as standard system components27 which includes open source software which is not a commercial product.
- Customization implies writing software code when the correct term should be configuration.
Right definitions; wrong place?
There are two definitions (hybrid system and automated script) in the New Chapter 4 glossary that should be included in Annex 11. Why are they in Chapter 4? Automated script cannot be found in the body of Chapter 4 because automatic validation script is used instead. Perhaps adding validation to the definition is appropriate.
The solution?
This is very simple; all defined terms should be in a single GMP glossary that is updated each time a new final chapter or annex is issued.
Summary
The updates of EU / PIC/S Annex 11 and applicable sections of Chapter 4 represent a major change and moved from interpretive to prescriptive regulation. This is compounded by the New Annex 11 not aligning with the current version (2011).
We have a bloated update with the following potential impacts:
Key sections from the existing A11 omitted etc.
- The application should be validated; IT infrastructure should be qualified.
- Relegation of Where a computerised system replaces a manual operation or system to the back of the principle – it should be at the front!
- No requirement for a system description or inventory of systems.
- Automated test tools and expansion to include any tools to aid validation.
- Incident management.
- Glossary problems, inconsistencies and omissions.
Limiting inspection flexibility?
- Could an inspection be reduced to a checklist instead of assessing a company on a flexible risk-based approach? This might be the case when inspecting IT security with the 20 clauses in section 15.
- Will inspectors be trained to inspect all areas covered by the New Annex 11?
More importantly, will they have technical expertise to apply during an inspection? - PIC/S PI-011 states in 23.4: … inspectors should use it selectively to build up a clear picture of a company's scale and complexity of on-site computerization (or automation) and investigate selectively the critical systems and risks.36 This flexibility is lost with the update.
- Furthermore, it is critical that there is a case-by-case risk assessment through an inspection.
Excruciating detail limits effective risk management
- All computerized systems are the same – except for the differences.
- It is the differences that require a flexible and risk-based approach to initial validation and on-going control.
- However, the overall content of the revised Annex 11 is far away from an effective risk-based approach.
Forget digital transformation: hybrid systems are OK!
- The Annex 11 concept paper6 mentioned digital transformation but the revision contains no significant expansion in this area.
- Moreover, there are 19 references to hybrid systems in Chapter 4 (WHY?) and New 13.9 presents a technically illiterate attempt to link handwritten signatures on with the underlying e-records.
- A great opportunity to push Pharma to improve has been missed.
Impact on regulated users
- Some companies that cannot or will not interpret a regulation may take the view if it is not stated – we don’t have to do this. These organizations star in FDA warning letters.
- Some companies cannot interpret regulations and say tell us how to be compliant. Doing nothing is not an option or excuse: talk to the regulators, read industry guides, e.g., GAMP 5 second edition (SE)27 attend training courses or engage a consultant to learn and implement compliant computerized systems
One-word summaries of the updates are:
- Prescriptive
- Bloated
- Repetitive
- Nit-picking
- Technically infeasible (OK, two words).
The authors appear to have forgotten the KISS principle (keep it simple, stupid). They should have only improved or clarified clauses where necessary. E.g. A11.10 has a good requirement for change control but nothing about configuration management, which would only have required a sentence to describe.

Understanding the DNA of a Unified Laboratory Informatics Platform
This Sapio Sciences whitepaper clarifies the difference between a platform and a product, advocating for unified, scalable informatics solutions across lab operations.View Whitepaper
It will be interesting to see the final version when issued, as we suspect that EMA / PIC/S will have to deal with an avalanche of adverse comments from all stakeholders.
Acknowledgements
We would like to thank, in alphabetical order, Monika Andraos, Markus Dathe, Jim Henderson, Bob Iser, Eberhard Kwiatkowski, Bunpei Matoba, Yves Samson (while on holiday!) and Paul Smith for their reviews and constructive comments that have improved our article. Great work in a very short timeframe and very much appreciated by us!



